“统计大讲堂”系列讲座第一百三十四讲顺利举行
2020-11-02
10月29日上午,“统计大讲堂”系列讲座第一百三十四讲举行。本次讲座采用在线直播的方式,讲座以“Scalable Integrative Statistical Inference for Whole-Genome Sequencing Association Studies”为主题,由哈佛大学副研究员李子林主讲。统计学院教师、应用统计科学研究中心研究员张波、李扬等参与讲座。统计学院教师孙韬主持本次讲座。
孙韬首先介绍了报告人的相关信息。李子林是哈佛大学陈曾熙公共卫生学院生物统计系副研究员,本科与博士毕业于清华大学数学科学系,主要研究方向为高维数据中的统计方法理论和遗传统计学,主持美国国家心肺血液研究所的基金一项,多篇学术论文在知名国际学术期刊发表。

李子林首先简单介绍了全基因组关联性分析的现状。在过去十几年中,全基因组关联研究(GWAS)广泛用于解析复杂疾病与数量性状的遗传结构。GWAS 已成功识别出数千种常见基因变异(common variants)。但是这些常见变异只能解释疾病遗传率中很小的比例。人类基因组中绝大部分的变异为罕见变异(rare variants),而且罕见变异对应的蛋白质更容易成为药物靶点。近年来,越来越多的全基因组测序研究(whole genome sequencing)正被采用,以发现与复杂性状和疾病相关的罕见变异。
GWAS中经典的检验方法是针对每个变异进行单独的检验,进而筛选出性状或疾病相关的变异。然而这种方法并不能直接应用于分析罕见变异。相比于单位点的检验,研究人员通常采用基于基因的多位点检验方法来分析罕见变异。不同于单独检测每个变异的效应,多位点检验识别的是一个基因或遗传区域当中多个变异的累积影响。如果一个区域中的多个变异与疾病或性状相关,那么基于多位点的检验可以大幅提高(统计)功效。同时多组学中的功能注释(functional annotation)提供了变异的生物学信息,有助于发现疾病与罕见变异之间的关联性。然而,现有的罕见变异关联分析方法尚未有效地利用这些变异的功能注释信息。
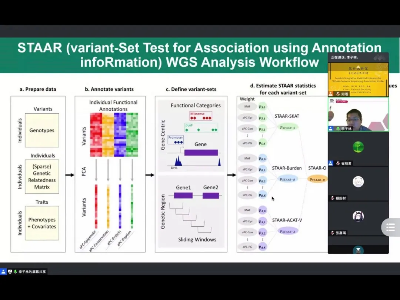
李子林所在的林希虹教授研究团队开发了基于多组学功能注释数据的罕见变异检验方法STAAR,有效地整合了变异多方面的生物学影响,进而显著地提高了罕见变异检验的统计功效。STAAR方法可以用于分析多种类型的表型数据,包括连续性表型和离散型表型,同时可以避免由人群分层现象和亲属及相关样本所带来的关联分析混杂性。李子林介绍了两种不同的方法来定义关联分析中的罕见变异集,一种是以基因为中心的分析(gene-centric analysis),另一种是以基于位置的遗传区段为单位的分析(genetic region analysis)。在以基因为中心的分析中,研究人员基于功能类别将基因中的罕见变异分为了5类,包括功能失活突变,错义突变,同义突变,启动子和增强子,并分别研究基因中每个类别与表型间的关联性。在以遗传区段为单位的分析中,研究人员采用预定长度的个碱基对(base pair)的滑动窗口,并通过每次将窗口“移动 一定碱基对来连续扫描整个基因序列。
李子林最后展示了STAAR方法在实际应用中的结果。研究人员将STAAR方法应用于TOPMed全基因组测序数据中,分析了30138人的四种脂质性状,新发现并验证了与低密度脂蛋白胆固醇(low-density lipoprotein cholesterol)相关的遗传位点,包括位于第7号染色体上基因NPC1L1中的破坏性错义突变(disruptive missense)和位于第19号染色体上基因APOC1P1下游附近的基因间区段中的滑动窗口。同时分析结果显示,与现有的罕见变异关联分析方法相比,STAAR通过有效地整合功能注释的信息显著地提高了检验的统计功效。
在提问交流环节,李子林耐心地解答提问,进一步阐释了模型在临床的实际应用以及在云计算平台的实现等。
此次讲座介绍的STAAR方法为大规模全基因组测序数据的罕见变异分析提供了快速,有效,准确的分析工具。此后统计大讲堂还将陆续推出多场精彩讲座,敬请关注。